Our long-standing collaboration with Prof. Stefan Krauss and Dr. Jo Waaler’s Cell Signaling and Drug Discovery research groups (Oslo University Hospital) has been a fruitful one. Symeres has contributed to the publication of three journal articles, three patents, and eight cocrystal structures of novel tankyrase inhibitors to date, and we continue to provide the project team with practical and intellectual expertise, creating novel molecules and high-quality scientific content, with great valorization potential.
A summary of the 2020 J. Med. Chem. publication can be found below.
Tankyrase 1 and 2 (TNKS1/2) are pivotal enzymes in the PARP family, influencing key proteins within the WNT/β-catenin and Hippo signaling pathways. Their inhibition offers a promising avenue for oncology therapeutics, as TNKS1/2 regulates the stability and activity of proteins like AXIN1/2 and AMOT, which affect cell proliferation and tumor growth. Although TNKS1/2 inhibitors have already been shown to display anticancer efficacy in mouse models against colorectal cancer, osteosarcoma, and melanoma (as monotherapy or in combination therapies), no TNKS1/2 inhibitors have been clinically approved to date. Hence, Prof. Stefan Krauss approached us to develop novel TNKS1/2 inhibitors with an improved physicochemical and biological safety profile, as these inhibitors showed potential for effective cancer treatment and valorization.
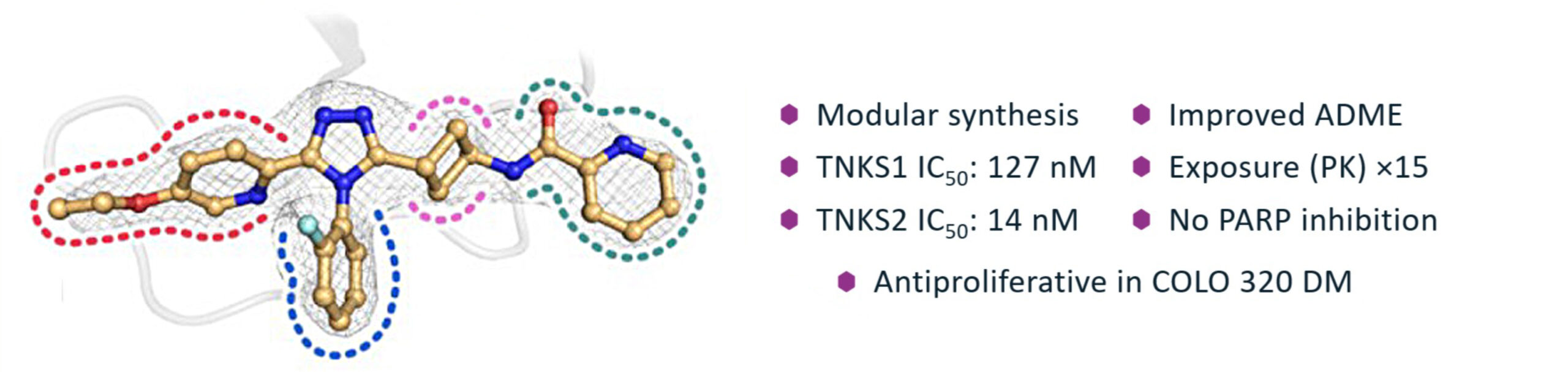
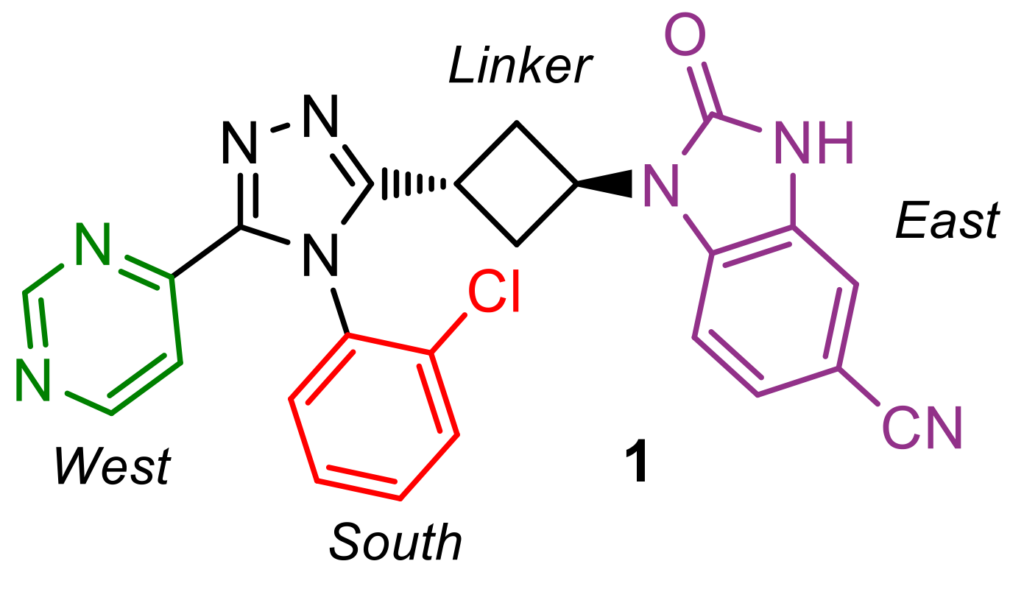
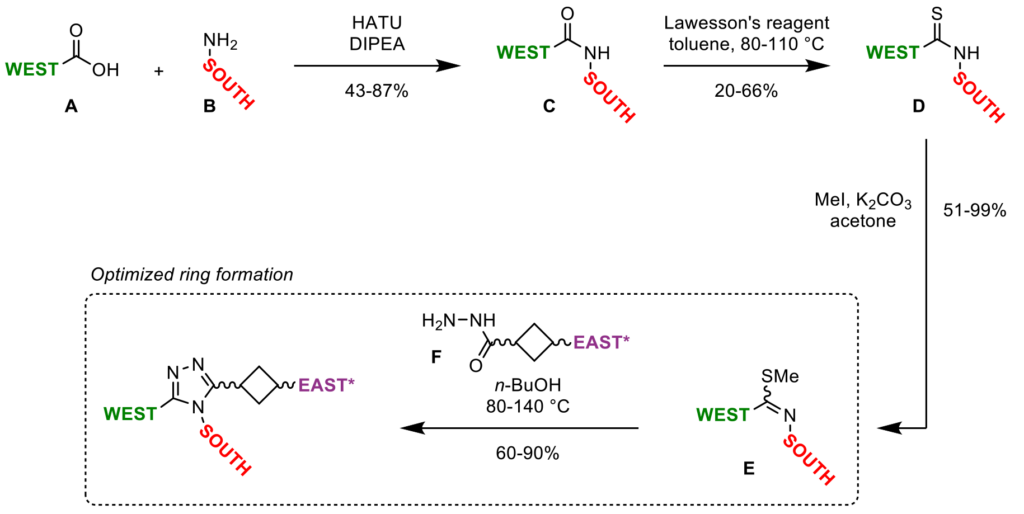
To optimize the ADME properties of our lead compound 1 (which had low aqueous solubility and a high Caco-2 efflux rate, Figure 1), while maintaining favorable potency and specificity towards TNKS1/2, our medicinal chemistry team devised a short synthetic route, which enabled rapid analog generation. Via HATU amide coupling, thionation, S-methylation, cyclization with a hydrazide linker, and subsequent deprotection/functionalization reactions, building blocks could be appended to the central core in a modular fashion (Scheme 1). This synthetic approach allowed for single-point modifications, enabling matched-pair analysis during the biological and physicochemical evaluation of these novel triazole analogs.
Highlighting Symeres’ synthetic excellence, our chemists optimized the key step in the triazole-core synthesis. While previously reported analogous triazole-ring formations required harsh conditions (TFA, DMAc, 120 °C), affording only poor to moderate yields (10–21%), we found that simply heating the mixture of iminothioether E and hydrazide F in 1-butanol at 80–140 °C significantly increased yields (60–90%), enabling a broader scope of South and West moieties on the triazole core structure (Scheme 1). Using these optimized reaction conditions, we provided our customer with over 105 substituted triazole analogs.
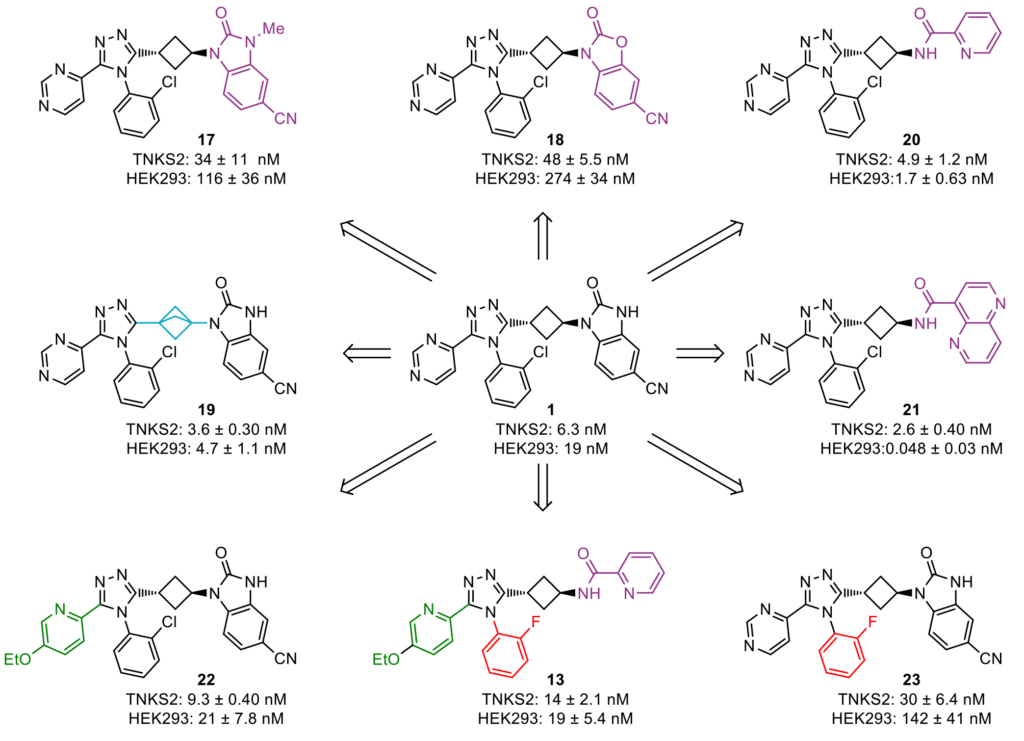
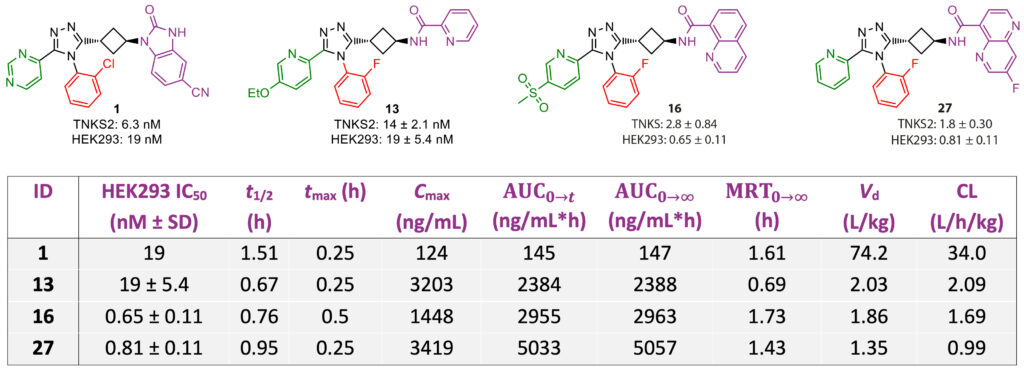
Biological evaluation of these triazole compounds in a TNKS2 biochemical assay and a WNT/β-catenin signaling pathway reporter assay in human HEK293 cells, based on luciferase activity, provided key insights into the structure–activity relationship of the chemical moieties appended on the central triazole core. These insights allowed our medicinal chemists to select promising compounds and further optimize their physicochemical properties, which eventually led to the discovery of lead molecule 13 (Figure 2).
During the lead-optimization study, two rotational isomers (atropoisomers) were observed for all compounds containing a South 2-chlorophenyl substituent. The rotation of this chemical moiety is hindered, due to steric hindrance by the West and East substituents. These atropoisomers could be analyzed and separated at Symeres’ Analytical Department viaSFC chromatography, with each atropoisomer displaying different retention times on a chiral column (Figure 3). Moreover, the two isolated atropoisomers of lead compound 1 showed a 30- to 60-fold difference in TNKS2 IC50 and HEK293 IC50 values, respectively.
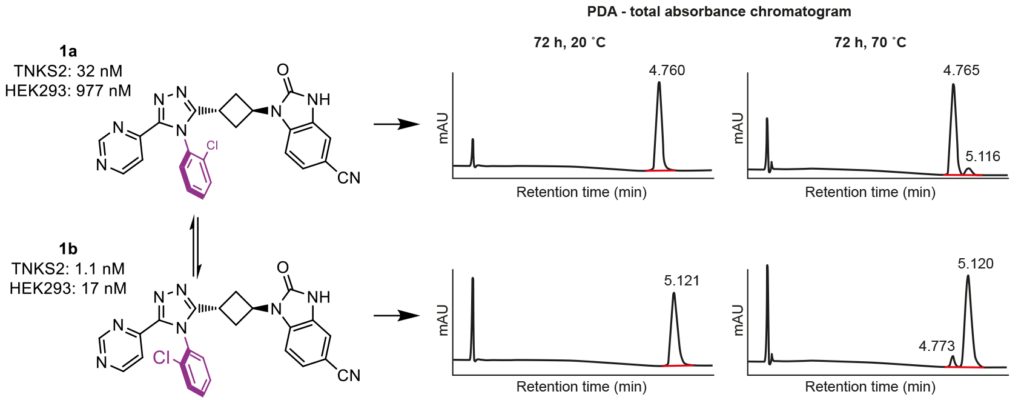
Since atropoisomers not only show different biological activities towards the desired target, but also different pharmacokinetic properties and off-target profiles, we sought another South moiety to prevent atropoisomerism. The 2-fluorophenyl group addressed this issue, showing no atropoisomerism, while occupying the same space as the 2-chlorophenyl group in the active site of TNKS2 (Figure 4A).
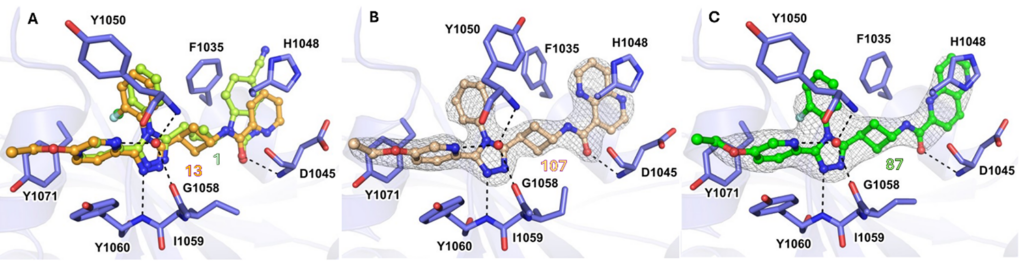
Cocrystal structures of tested inhibitors with TNKS2, critically analyzed by our computational chemists, were instrumental in understanding the influence of atropoisomerism on the binding mode, showing a preferred conformation of the 2-halophenyl group in the TNKS2 active site. While an unsubstituted South phenyl analog showed a similar orientation to that of the 2-halophenyl analogs in a cocrystal, the potency decreased significantly; this was attributed to conformational changes of the Tyr1050 residue, which covered the active site (Figure 4B). Although larger 1,5- and 1,6-naphthyridine East moieties were found to display a more efficient π−π-stacking interaction with His1048 and a hydrophobic interaction with Phe1035 (Figure 4C), these naphthyridine derivatives showed lower microsomal stability compared to pyridine East analogs. These stability issues were addressed by the introduction of a fluorinated 1,5-naphthyridine ring, or by swapping the naphthyridine moiety for a quinoline, yielding compounds 16 and 27, which both showed nanomolar IC50values for TNKS2 and HEK293. Compared with our client’s initial lead compound 1, all three optimized analogs (13, 16, and 27) showed significantly improved cellular efficacy and pharmacokinetic profiles in mouse models, displaying 15–35-fold higher exposure levels, combined with a lower volume of distribution and clearance (Table 1).
Administration of 13 to COLO320DM cancer cells, which contained WNT/β-catenin signaling-inducing APC mutations, clearly illustrated tankyrase inhibition, reducing protein levels of TNKS1/2, downstream-stabilized AXIN1/2 proteins, and non-phosphorylated β-catenin in both the nuclear and cytoplasmic fractions. Transcription of target genes of the WNT/β-catenin signaling pathway, AXIN2, DKK1, APCDD1, and NKD1, also decreased upon treatment with 13. Ultimately, exposure to 13 decreased the viability and proliferation of COLO320 DM cells, while wild-type APC RKO colorectal cancer cells were only modestly affected by treatment at 10 µM concentration.
Due to the promising results obtained in this first lead optimization, the team at Oslo University Hospital was keen to continue collaborating, which yielded an even more attractive compound that will be described in a follow-up publication in due course.
To access the J. Med. Chem. article, go to https://pubs.acs.org/doi/10.1021/acs.jmedchem.0c00208.
Want to explore how Symeres can support and accelerate your academic research? Have a look at our Services page to see what we can offer: https://symeres.com/services/.
Do you prefer a more personal conversation? Don’t hesitate to contact our co-author and Director of Medicinal Chemistry, Anita, with any questions (anita.wegert@symeres.com); at Symeres, we’re always happy to help you out!
