Home / News
Our news


Symeres and Axxam Appoint Russell Thomas as Chief Scientific Officer to lead their collaborative integrated discovery platform
Symeres and Axxam have appointed Russell Thomas, Ph.D., as Chief Scientific Officer, reinforcing the company's scientific leadership and integrated CRDMO offering. Russell returns to Symeres with extensive experience spanning drug discovery and development, helping to drive the company's next phase of growth.

10.06.26
Symeres expands spray drying capabilities to accelerate complex small molecule development
At Symeres, we are pleased to announce the expansion of our integrated spray drying capabilities at our Cranbury, New Jersey CMC development site. This investment strengthens our ability to support formulation development for poorly soluble and development-challenged small molecule drug candidates, helping biotech and pharmaceutical companies accelerate progress from preclinical development through to Phase II clinical studies.

13.05.26
Symeres featured in Speciality Chemicals: navigating biotech’s new reality
Symeres has been featured in Speciality Chemicals discussing how biotech companies are adapting to tighter funding conditions, rising development complexity and increasing pressure to demonstrate value earlier in development. In the article, Philip Payne and Dr Goran Verspui explore how integrated scientific expertise, AI-enabled optimisation and connected development workflows are helping biotechs accelerate progress while reducing risk.

06.05.26
Symeres appoints Henning Steinhagen as CEO to drive next phase of strategic growth
Symeres welcomes Dr. Henning Steinhagen as Chief Executive Officer, bringing over 25 years of leadership experience across pharma, biotech and the CRO/CDMO sector. His appointment marks an exciting step forward as Symeres continues to strengthen its integrated “One Symeres” platform and support drug discovery and development from concept to clinic.
31.03.26
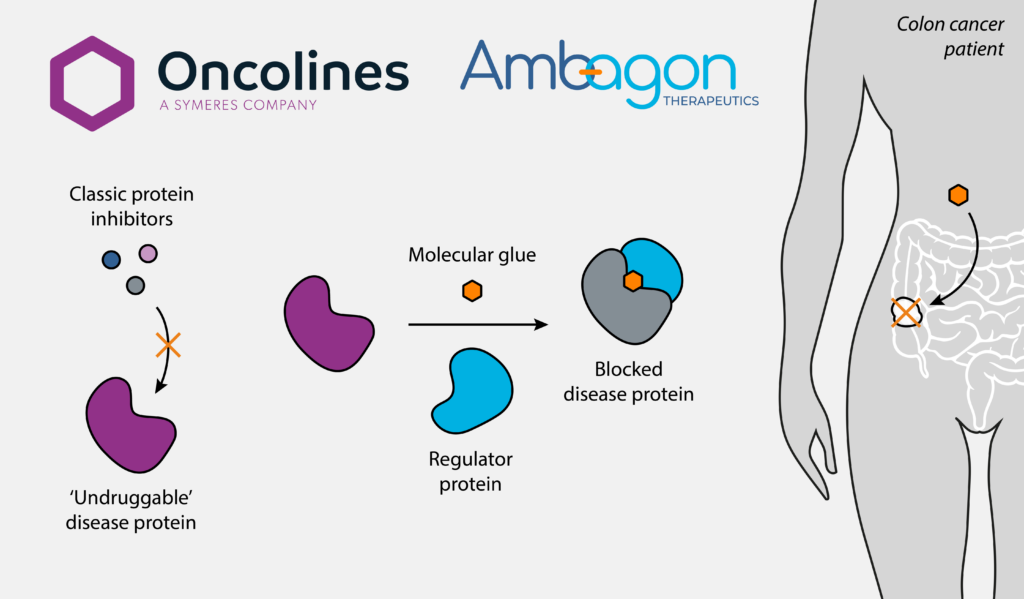
Symeres collaborates with Ambagon Therapeutics on molecular glue research in colorectal cancer
Symeres has partnered with Ambagon Therapeutics to evaluate molecular glues for colorectal cancer, combining translational research and in vitro studies to support early-stage development.

03.02.26
What are the top drug development trends for 2026? Exemplify CSO Paul O’Shea discusses what to watch
Exemplify BioPharma CSO Paul O’Shea shares his perspective on the key drug development trends for 2026, including AI-driven decision-making and drug repurposing strategies.

17.12.25
Symeres earns first SBTi approval as part of long-term sustainability journey
Nijmegen, Netherlands – 17.12.25 – Symeres, a leading contract research, development, and manufacturing organisation (CRDMO), has received its first approval from the Science Based Targets initiative (SBTi) for its greenhouse gas (GHG) emissions reduction targets. This marks a major milestone in Symeres’ environmental strategy and affirms its commitment to building a more sustainable, low-carbon future. […]

10.09.25
Symeres announces acquisition of DGr Pharma
DGr Pharma specializes in preclinical and clinical regulatory strategy and consultancy for biotech and pharmaceutical partners working in early drug development. Its core services include chemical-pharmaceutical, non-clinical and clinical development planning, quality assurance, and regulatory submissions. DGr Pharma has deep expertise in small and large molecules including antibodies, ADCs, and oligonucleotides.

03.06.25
Symeres and Yoneda Labs use AI to optimize cross-coupling reactions
San Francisco, May 29, 2025 – Yoneda Labs, a leader in computational tools for reaction optimization, has successfully collaborated with Symeres, a leading transatlantic, small molecule contract research and manufacturing organization. The aim was to optimize transition metal catalyzed cross-coupling reactions to improve reaction yield and efficiency utilizing Yoneda Labs’ cutting-edge software.

16.12.24
New production lines D1 and D2 at our Prague site
The new production lines D1 and D2 were launched for the first time in May 2024 at the Prague site. The first batch of 11 kg of API was produced in the following weeks, and the tremendous efforts of the many people involved in planning the entire production line have finally paid off. Since the production line was launched, all equipment has been continuously utilized.

06.12.24
New NMR technology – meet “Maggie”
Nuclear Magnetic Resonance (NMR) Spectroscopy is a powerful analytical tool well known to all organic chemists and to anyone that works in pharmaceuticals. By placing a sample dissolved in deuterated solvent into a strong magnetic field (roughly 4000x the strength of Earth’s magnetic field) and pulsing it with radio frequencies, the chemist is given an enormous amount of information from the identity and placement of functional groups to relative stereochemistry of diastereomers. Recently, Exemplify added NMR to our expanding list of capabilities.