In our recent ChemMedChem publication, colleagues from our Drug Discovery Department, in collaboration with WeLab Barcelona, reported the synthesis and biological activity of novel pain therapeutics, which target both the α2δ-1 subunit of voltage-gated calcium channels (Cavα2δ-1) and the μ-opioid receptor (MOR). A short summary of this publication can be read here.
Unfortunately, pain is an unavoidable part of human existence, and humans have been using pain therapeutics for centuries. However, despite all the innovations and research in the field, current pain treatment options still suffer from suboptimal efficacy, unwanted side effects, withdrawal symptoms, and tolerance. Therefore, the need for novel pain-relieving drugs remains high, as, in addition to the issues listed above, the condition still poses a high socioeconomic burden.
Pain is mediated via several signaling pathways, and these pathways are often addressed by administering a combination of multiple pain therapeutics, which target multiple mediators at once. These drug combinations can work synergistically, allowing for pain relief at lower doses and, thereby, reducing the risk of adverse effects. However, it is less practical for a patient undergoing treatment to keep up with multiple dosing schedules for different drugs, while drug combinations also present a potential risk of drug–drug interactions and require more intensive pharmacokinetic profiling. Hence, a rationally designed single compound with multimodal activity is considered to tackle these issues, aiming to provide better efficacy, improved patient compliance, simpler pharmacokinetics, a reduced risk of drug–drug interactions, and less variability between patients.
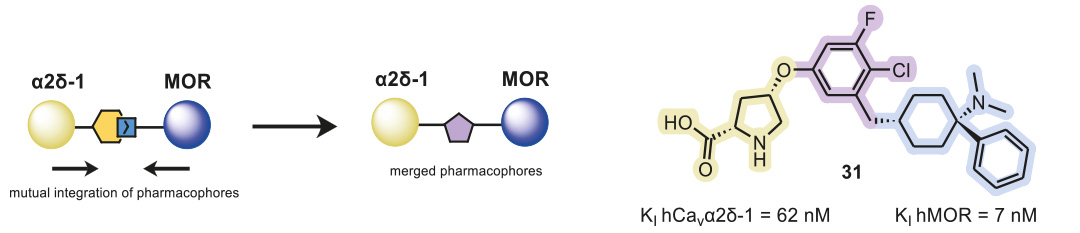
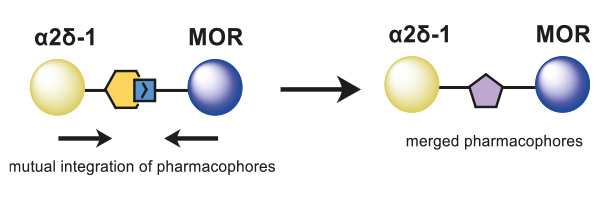
The overlapping and merging of two known pharmacophores of the Cavα2δ-1 subunit and MOR enabled the design of novel small-molecule inhibitors as dual ligands, which displayed binding elements for both targets on a central core structure (Figure 1). Following this approach, MOR-binding structural features were added to a previously published Cavα2δ-1 ligand, and via the synthesis of a small, focused, analog library, the structure–activity relationship of these hybrid binders was explored.
Showcasing Symeres’ accumulated synthetic experience, these dual ligands could be obtained from readily available starting materials in less than five steps, via well-established synthetic methodologies, such as Mitsunobu reactions, reductive aminations, and Suzuki–Miyaura cross-coupling chemistry. This allowed for rapid analog generation, supplying our customer with a steady influx of new molecules, while providing them with the flexibility to prioritize and pivot syntheses in short time frames.
Through exploitation of Symeres’ strong network of collaborating academic institutions, single-crystal X-ray diffraction (XRD) was performed at Radboud University, which allowed for the definitive assignment of the stereochemistry of the ligands, some of which contained up to four chiral centers. Combining XRD data with in silico docking studies, performed by Symeres’ Computational Chemistry team, provided our client with key insights into the stereoselective preferences of the biological targets and binding mode of the ligands. After fine-tuning the ligand’s affinity for both Cavα2δ-1 and MOR targets, monitored via radioligand binding assays (gabapentin for Cavα2δ-1, DAMGO for MOR) and cAMP assays (MOR), the most promising molecule was chosen for more intensive pharmacological profiling.
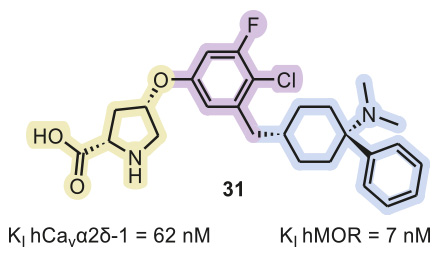
Although one may not expect permeation of the blood–brain barrier by a polar molecule (elogD7.4 = 1.0) with a relatively high molecular mass (475 Da), the amino acid derivative 31 (Figure 2) displayed significant brain concentrations upon intraperitoneal injection and oral administration.
Disubstituted pyrrolidine 31 showed no activity in a selectivity panel of off-targets associated with undesirable side effects and other pain targets (including the human ![]() -1 receptor, serotonin receptors and transporters, adrenoreceptors, the histamine H1 receptor, and dopamine and norephinephrine transporters), except the δ-opioid receptor DOR (Ki=195
-1 receptor, serotonin receptors and transporters, adrenoreceptors, the histamine H1 receptor, and dopamine and norephinephrine transporters), except the δ-opioid receptor DOR (Ki=195![]() 22 nM). Furthermore, no inhibition was observed for 31 in CYP screening and hERG ion-channel assays, nor did the compound show in vitro cytotoxicity in neutral red uptake assays and MTT assays in HepG2 cells at 100 µM. Additionally, compound 31 showed no activity in the Ames bacterial mutation assay, indicating its lack of mutagenicity.
22 nM). Furthermore, no inhibition was observed for 31 in CYP screening and hERG ion-channel assays, nor did the compound show in vitro cytotoxicity in neutral red uptake assays and MTT assays in HepG2 cells at 100 µM. Additionally, compound 31 showed no activity in the Ames bacterial mutation assay, indicating its lack of mutagenicity.
In various rodent models of pain, including thermal and mechanical stimuli, as well as long-lasting pain, compound 31demonstrated an analgesic effect, with maximum efficacy achieved in the formalin test and paw-incision model.
Additionally, it was observed that compound 31 did not induce tolerance with repeated administration, unlike commonly used opioids, such as morphine. Furthermore, withdrawal signs were not evident after treatment with compound 31, indicating a potentially favorable profile compared to opioid drugs.
Want to find out more about how Symeres’ highly motivated team of synthetic, medicinal, and computational chemists can help you drive your drug discovery projects forward? Have a look at our drug discovery page at https://symeres.com/services/drug-discovery/.
To access the ChemMedChem article, go to https://chemistry-europe.onlinelibrary.wiley.com/doi/epdf/10.1002/cmdc.202300473.
Don’t hesitate to contact our Director of Medicinal Chemistry, Anita, with any questions (anita.wegert@symeres.com); at Symeres, we’re always happy to help you out!
